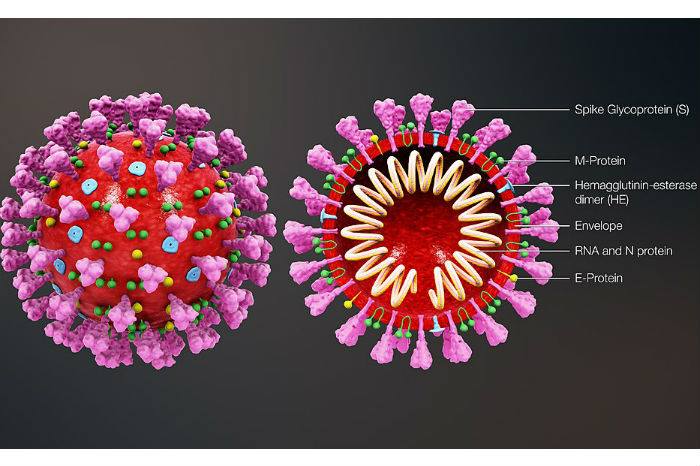
El SARS-CoV-2, de la familia de los coronavirus, tiene por dentro una cadena de ARN, que al igual que el ADN en nuestras células, tiene información. En este caso, toda la información necesaria para poder hacer copias del virus. Cuando infecta la célula, podemos decir que el virus "toma de rehén" a las proteínas que necesita para poder reproducirse "obligando" a la célula a leer la información que trae en su ARN.
Además, el virus tiene una capa de lípidos (grasas) que lo recubre (esa es la capa que rompemos cuando nos lavamos las manos con jabón), y unas proteínas que fueron bautizadas SPIKE ("pinches") que le dan el aspecto tan característico a los coronavirus (sí, de ahí viene lo de "corona").
Desde que comenzó a extenderse el virus y tomamos conciencia de la importancia de lidiar con la pandemia, se orquestó a lo largo del mundo una respuesta sin precedentes. Científicos de todas partes comenzaron a colaborar para comprender el funcionamiento del virus, cómo enferma a las personas y qué podemos hacer para frenarlo.
Uno de estos esfuerzos colectivos consiste en la secuenciación de genomas virales.
¿Qué es secuenciar un genoma?
Secuenciar el genoma del virus es simplemente conocer la cadena de ARN viral entera. Su secuencia a simple vista es algo que no parece tener sentido, algo así como GCAAAAUCGUUAAAAGGC......... etcétera. "Secuenciar un genoma" significa tener esa información completa para todo el ARN (o ADN) de un organismo.
La primera vez que se secuencia el genoma de un organismo es la más complicada, porque en principio se sabe muy poco de esa cadena. Lo que se hace es juntar mucho ADN / ARN de la muestra, partirlo en pedacitos (lecturas o “reads”) de distinto largo y leerlos. Si hay suficiente ADN / ARN, muchas de esas lecturas se van a solapar, es decir, una va a terminar con la misma secuencia con que empieza la siguiente, y así se puede ir armando la cadena completa en orden.
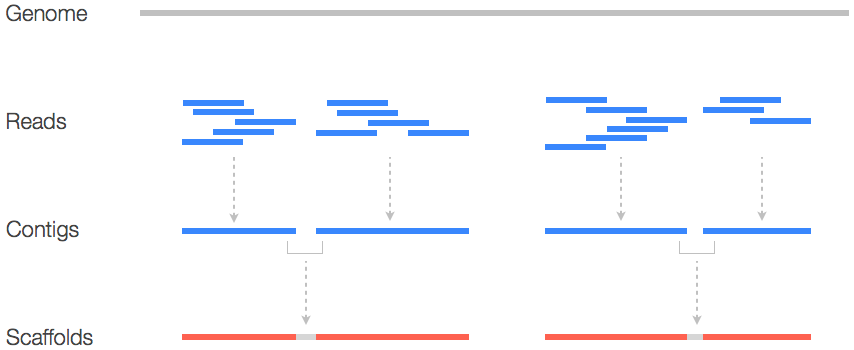
El genoma completo del SARS-CoV2 se secuenció por primera vez con muestras tomadas de pacientes de Wuhan, China (donde comenzó la pandemia). Desde entonces, se hizo lo mismo con muestras de muchas partes del mundo.
Una vez que se tiene un genoma completo secuenciado por primera vez, analizar otras muestras es relativamente más fácil, porque esa primera secuencia se usa como genoma "de referencia". Es como cuando queremos resolver un rompecabezas y tenemos la foto en la tapa: podemos mirar la pieza que tenemos en la mano y ver dónde encaja en la foto de referencia. Con las secuencias es igual: secuenciamos muchos pedacitos pequeños y vemos a qué parte del genoma de referencia se parecen, y así vamos armando cada nuevo genoma en base a cada muestra diferente. Todas las secuencias del SARS-CoV2 se encuentran a disponibilidad del público en bases de datos.
¿Para qué secuenciar un genoma?
Conocer la secuencia es importante porque permite entender qué está haciendo dentro de las células. Podemos encontrar algunas partes que son similares a otros virus que conocemos mejor y deducir qué significa esa información. A veces esa secuencia corresponde a una proteína importante para el virus, y eso puede servirnos para desarrollar fármacos que le impidan funcionar.
¿Para qué secuenciar VARIOS genomas?
¿Por qué secuenciar varios genomas en lugar de quedarnos con uno solo y listo? Al igual que pasa con el ADN, el ARN puede sufrir errores en su replicación, que conocemos como mutaciones. Es decir que una muestra tomada en un lugar puede tener diferencias en su secuencia con respecto al genoma de referencia. En nuestra metáfora del rompecabezas, cada pieza se va a parecer mucho a la foto de referencia, pero va a tener tal vez pequeñas modificaciones. Esas diferencias entre el genoma de referencia y cada nueva muestra son las mutaciones.
Cuando se reproducen, los virus mutan de forma aleatoria. Esto ocurre relativamente rápido, especialmente en los que tienen ARN. En algunos virus “suertudos” pueden aparecer mutaciones en regiones que nuestro sistema inmune ya reconocía, y eso hace que puedan "burlar" a nuestras defensas, aún cuando ya estuviésemos inmunizados. Eso pasa con la gripe: uno puede vacunarse, pero la cepa del año siguiente es tan distinta que hace falta una nueva vacuna, porque los anticuerpos generados en la vacunación del año anterior no la reconocerían. Por eso la gripe vuelve como oleadas de año a año y puede infectarnos varias veces, a diferencia de otras enfermedades para las que nos hemos inmunizado de por vida.
Secuenciar muchos genomas del SARS-CoV2 de distintas partes del mundo sirve para prever qué va a pasar a futuro con los fármacos o las vacunas. ¿Podemos diseñar una única vacuna que sirva para todos? ¿Será como la gripe que año a año cambia y necesitamos nuevas vacunas?
Lo que sabemos por ahora sobre el SARS-CoV2 es que aparentemente no muta tan rápido como otros virus de ARN. Algo que se descubrió al secuenciar su genoma es que este virus tiene una proteína que corrige errores de replicación. Eso en realidad es buena noticia para nosotros, porque significa que el virus muta más lentamente que otros, y si sigue siendo así, un único tratamiento o vacuna podría servir para todos los pacientes. Por ahora, lo importante es que en principio ninguna de las mutaciones que acumuló afectaría la forma de las proteínas SPIKE que debería reconocer una eventual vacuna. O sea: por ahora alcanzaría con diseñar una única vacuna. Y lo mejor es que, si esto se mantiene, no tendríamos nuevas “oleadas” de COVID-19 que pudiesen volver a infectar a los ya curados.
Además, los métodos de diagnóstico también se basan en conocer la secuencia de algunas partes del genoma viral. Es decir que conocerlas nos permite mejorar los diagnósticos y bajar la tasa de errores.
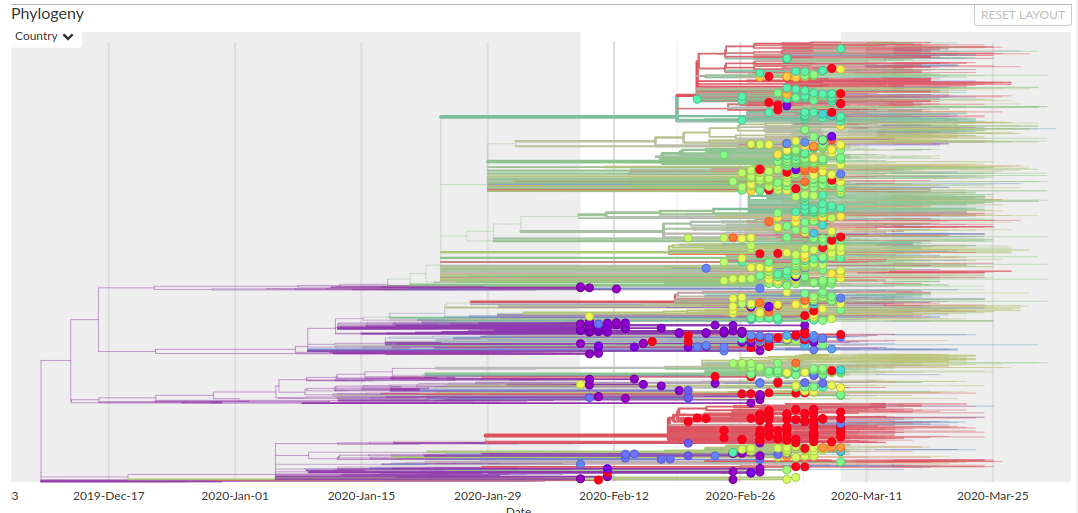
Por último, secuenciar muchos genomas nos permite "rastrear" cómo se fue extendiendo el virus a lo largo del mundo. Se puede ir armando una filogenia, una especie de "árbol genealógico" del virus en base a las diferencias entre las muestras y así se puede saber más o menos qué camino recorrió la infección a lo largo del mundo:
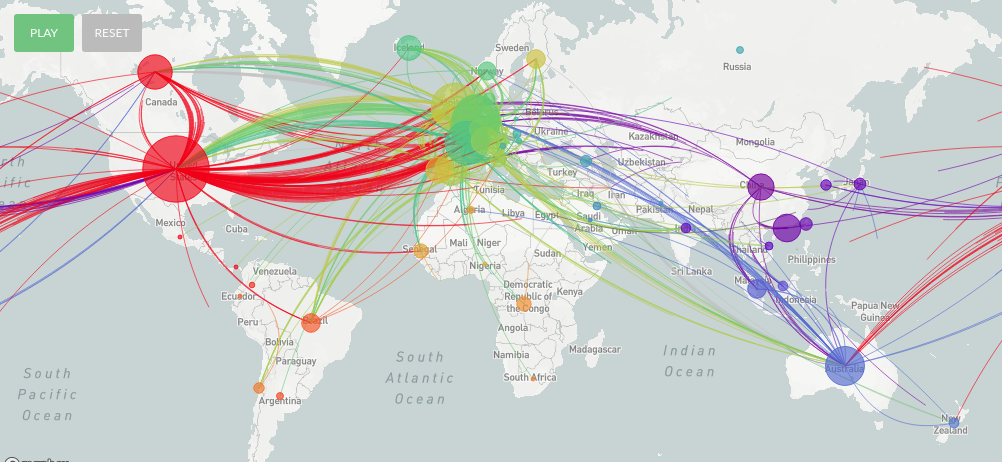
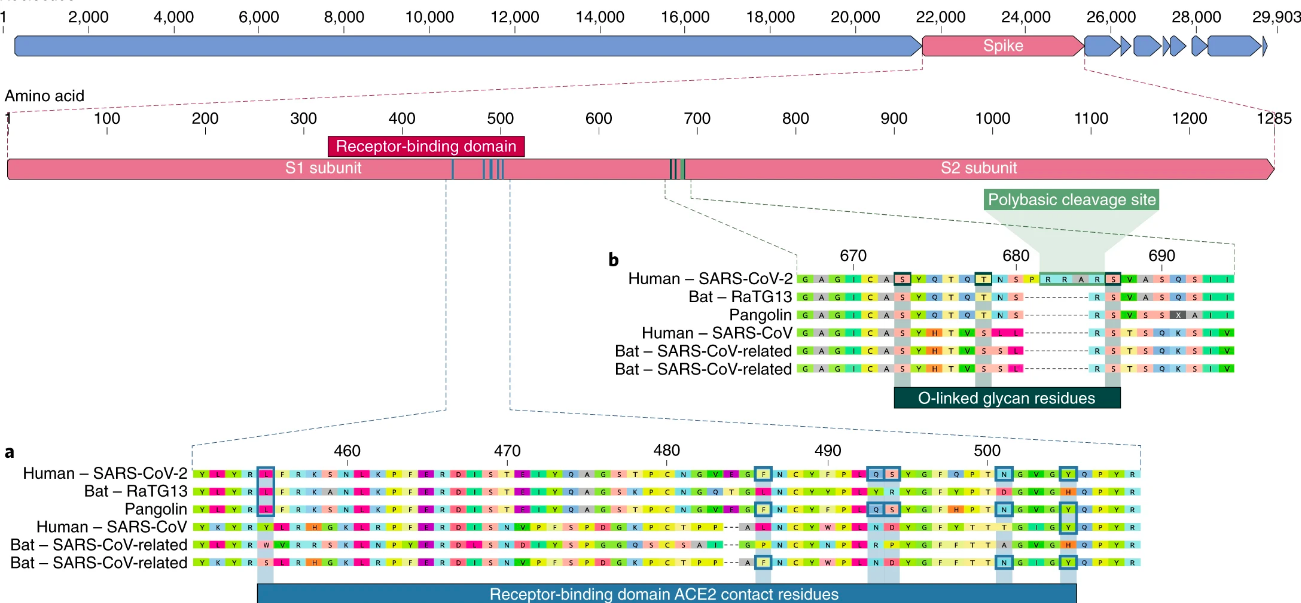
Esto se puede usar también para encontrar el origen del virus. Todos leímos acerca de la sopa de murciélago o los pangolines. Esto (que no es más que tratar de echarle la culpa de la pandemia a costumbres de otras personas sin mucho motivo) es porque en poblaciones de ambos animales se encontraron coronavirus muy parecidos en su secuencia al SARS-CoV2, aunque aún los científicos no están seguros de cuál de los virus fue el que comenzó a infectar a humanos ni cómo se produjo este "salto".
¿Qué nos dicen los tres genomas argentinos?
Finalmente, pasamos a la noticia de ayer. En el Malbrán se secuenciaron tres genomas "argentinos" (es decir: se analizaron tres muestras de virus provenientes de pacientes argentinos, independientemente de dónde se hayan contagiado).
Por ahora, lo que sabemos de estas muestras es que una de ellas es similar las encontradas en pacientes europeos, otra a la correspondiente a pacientes de EEUU, y la tercera similar a las secuenciadas en Asia, aunque el paciente argentino se habría contagiado en Europa de una persona que había estado en el continente asiático. Además estas secuencias no tienen muchas mutaciones con respecto a estas muestras, lo que es una buena noticia.
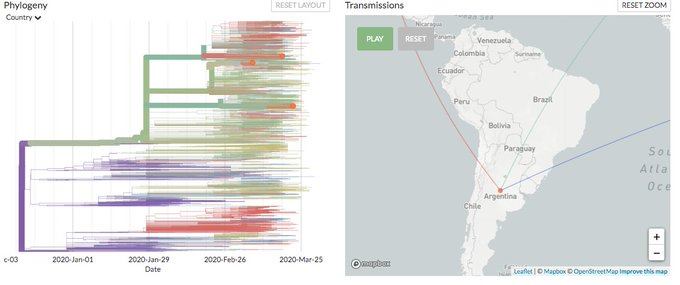
Más adelante seguramente tendremos más genomas "argentinos" secuenciados, una tarea que es lenta, insume mucho trabajo y recursos. Eso nos permitirá seguir rastreando cómo se extendió la infección en nuestro país y también colaborar en las investigaciones que se están llevando a cabo alrededor del mundo para lidiar con la pandemia.
